


Una investigación coordinada por la U746 CIBERER que lidera Belén Pérez en el CEDEM-CBM-UAM ha identificado 20 casos de hiperfenilalaninemia (HFA) con defectos en el gen DNAJC12, recientemente descrito. La HFA es una condición metabólica genéticamente heterogénea que se detecta desde hace más de 50 años en las pruebas de cribado neonatal. Defectos en este gen también se han relacionado con la enfermedad de Parkinson de presentación temprana.
Los autores del estudio, publicado en Human Mutation, proponen que la confirmación del diagnóstico del cribado de hiperfenilalaninemia incluya la secuenciación de DNAJC12, lo que permitirá la prescripción de un adecuado tratamiento. Además, recomiendan la inclusión de este gen en paneles genéticos de diagnóstico de Parkinson.
La U746 es un centro de referencia en el diagnóstico genético en España de enfermedades metabólicas entre las que se encuentra la HFA, lo que le ha permitido realizar este estudio retrospectivo de casos con HFA sin confirmación genética. El trabajo es el resultado de una colaboración multidisciplinar en la que ha participado la U703 CIBERER liderada por Rafael Artuch en el Hospital Sant Joan de Déu de Barcelona, centros hospitalarios de otras ciudades españolas (Valencia, Sevilla, Madrid, Zaragoza, Valladolid y Murcia) e investigadores de Chile y Noruega.
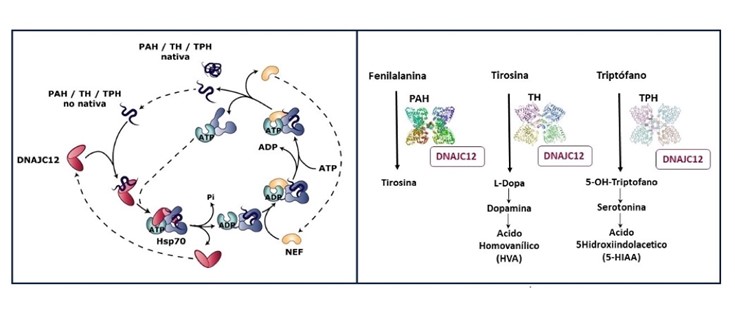
El 98% de los pacientes con HFA son portadores de variantes bialélicas en el gen de la fenilalanina hidroxilasa (PAH), que dan lugar a la fenilcetonuria (PKU), mientras que el 2% son portadores de variantes en genes implicados en la síntesis o regeneración de la tetrahidrobiopterina (BH4), cofactor de la PAH y de la tirosina (TH) y triptófano hidroxilasas (TPH), estas dos últimas implicadas en la síntesis neuronal de dopamina y serotonina respectivamente.
El gen DNAJC12 descrito en los 20 casos analizados codifica para una cochaperona que asiste al plegamiento de las tres hidroxilasas. Mutaciones en este gen también han sido relacionadas con Parkinson de presentación temprana. Se han identificado 4 variantes no descritas, siendo una de ellas p.Trp175Ter, muy frecuente en los pacientes y también, sorprendentemente, muy frecuente en la población española según los datos del SpanishVariantServer (0.3%).
Los estudios de genómica funcional han revelado que la variante p.Trp175Ter afecta tanto al plegamiento de la PAH como de la TH pero no afecta al plegamiento de la TPH. Atendiendo a estos resultados se ha propuesto a los clínicos una revisión del tratamiento de estos pacientes, que estaban clasificados como defectos en PAH, con el propósito de valorar la posibilidad de una administración de precursores de dopamina que eviten secuelas neurológicas.
Además, se han realizado estudios de coexpresion de variantes de plegamiento descritas en PAH con DNAJC12. Los resultados sugieren que DNAJC12 podría ser un modificador del fenotipo PKU además de una nueva diana terapéutica.
Artículo de referencia:
Pathogenic variants of DNAJC12 and evaluation of the encoded cochaperone as a genetic modifier of hyperphenylalaninemia. Gallego D, Leal F, Gámez A, et al. Human Mutation. 2020;1–10. https://doi.org/10.1002/humu.24026
(*) Explicación de la figura: Papel de DNAJC12 en el plegamiento de las hidroxilasas PAH, TH y TPH. Izquierda: Ciclo de plegamiento de las hidroxilasas por la chaperona Hsp70. DNAJC12 ejerce de cochaperona guiando la especificidad del proceso. Derecha: Reacciones de las hidroxilasas PAH, TH y TPH.




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




