



Las investigadoras María Clemente, Mónica Fernández, Núria Camats y Laura Audí, de la U712 CIBERER que lidera el Prof. Antonio Carrascosa en el Vall d’Hebron Institut de Recerca, han participado en un estudio multicéntrico que ha caracterizado un nuevo gen causante de un nuevo síndrome en pacientes con patología suprarrenal y renal. El estudio ha sido dirigido por la Dra Louise Metherel, del Queen Mary University of London, y se ha publicado recientemente en el Journal of Clinical Investigation.
El gen sphingosine-1-phosphate lyase (SGPL1) se ha identificado como causa de afectación multiorgánica, particularmente de la glándula suprarrenal (insuficiencia suprarrenal primaria) y del riñón (síndrome nefrótico corticorresistente). Este síndrome podría representar una nueva enfermedad de almacenamiento excesivo de lípidos, similar a las enfermedades de Niemann-Pick y de Fabry. Estas enfermedades son multisistémicas y progresivas, muchas veces con implicación neurológica.
 |
|
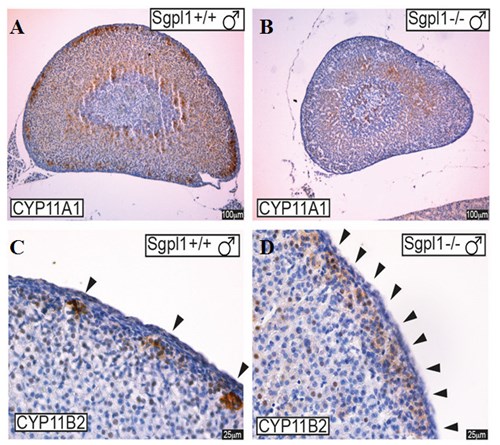
Fig. 1. Histología de la glándula suprarrenal. Déficit de expresión de enzimas de la esteroidogénesis CYP11A1 y CYP11B2 en ratones sin Sgpl1 (Sgpl1–/– ). (A, B) La enzima CYP11A1 (cholesterol side-chain cleavage) se expresa menos intensamente. (C, D) La enzima CYP11B2 (aldosterone synthase) no presenta la expresión moteada característica.
|
En el estudio participaron pacientes de Reino Unido, Turquía, Paquistán, Perú y España (Hospital Vall d’Hebron), de los cuales se analizó el DNA (mayoritariamente por secuenciación masiva de exoma), en el que se detectaron mutaciones en el gen SGPL1. No todos los pacientes tienen los mismos síntomas: hay pacientes con predominio de patología renal y otros con afectación suprarrenal. Además, la enfermedad parece progresiva y algunos pacientes han ido perdiendo capacidades motoras y cognitivas con el tiempo. De hecho, este gen también se ha relacionado con la neuropatía de Charcot-Marie-Tooth en otro grupo de pacientes. Según los investigadores, el diagnóstico genético de estos pacientes permitirá un tratamiento correcto, consejo genético y un seguimiento minucioso para detectar rápidamente nuevos síntomas.
Particularmente, la paciente procedente del Hospital Vall d’Hebron presentó síndrome nefrótico corticorresistente el primer año de vida y ha necesitado dos trasplantes de riñón. Más adelante, se le detectó insuficiencia suprarrenal primaria, ictiosis y un hipotiroidismo primario no autoinmune. Es portadora de una mutación en homocigosis al inicio del gen SGPL1. Dos familiares también están afectados: uno con síndrome nefrótico y el otro con las dos patologías. La insuficiencia suprarrenal primaria en la paciente consiste en un defecto en la producción de hormonas esteroideas suprarrenales (glucocorticoides y andrógenos) y se manifestó con hiperpigmentación, hipoglucemia e hipotensión. Algunos pacientes presentan formas parciales sin déficit de mineralocorticoides. La insuficiencia suprarrenal primaria puede ser fatal si no se detecta, puede manifestarse asociada a otros trastornos y puede tener un origen genéticamente heterogéneo.
El mismo estudio resalta la importancia de la ruta de los esfingolípidos en la función suprarrenal y renal y sugiere una participación potencial en otros tejidos como el cerebro y la glándula tiroidea. Ratones con el mismo defecto genético (Sgpl1–/–) presentan las mismas principales características de la enfermedad en humanos y podrían ser un buen modelo para ensayar nuevos tratamientos para esta grave patología.
Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. Prasad et al. Journal of Clinical Investigation. 2017 Mar1;127(3):942-953.




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




