



Un nuevo estudio multicéntrico liderado por José María Bastida, de la Unidad de Trombosis y Hemostasia del Hospital Universitario de Salamanca – IBSAL, Alessandra Balduini, de la Universidad de Pavia (Italia), y José Rivera, de la U765 CIBERER que coordina Javier Corral en el IMIB, ha descubierto el mecanismo de patogenicidad que causa la trombocitopenia hereditaria relacionada con GALE y su papel esencial en la glicosilación de proteínas plaquetarias.
Las trombocitopenias hereditarias constituyen un grupo heterogéneo de enfermedades raras caracterizadas por una disminución del número de plaquetas en la sangre, causadas por mutaciones germinales en genes clave para la formación de las plaquetas. Algunos pacientes con estas enfermedades son prácticamente asintomáticos, pero otros cursan con hemorragias clínicamente relevantes, e incluso trastornos graves en distintos órganos o tejidos, como un riesgo alto de desarrollar neoplasias hematológicas, problemas cardiacos o neuromusculares.
La trombocitopenia relacionada con GALE causa una disminución en la actividad de la enzima UDP-Galactosa 4-Epimerasa. Esta enzima está involucrada en el metabolismo de la galactosa, por lo que la forma general de la enfermedad es asintomática, y la única manifestación evidente es la galactosemia, es decir, un incremento de los niveles de galactosa en sangre. Sin embargo, existen formas muy severas y poco frecuentes de la enfermedad, en el que los pacientes manifiestan retraso cognitivo, alteraciones cardiacas, problemas hepáticos, tales como unos elevados niveles de bilirrubina, o desarrollo de cataratas a una edad muy temprana.
En el nuevo trabajo, que acaba de ser publicado en la prestigiosa revista Blood, se han descrito dos familias no relacionadas con una manifestación sindrómica grave, sangrado moderado, y trombocitopenia muy grave con la presencia de plaquetas gigantes en sangre.
Los autores del trabajo han demostrado que esta alteración se debe a una alteración en los megacariocitos de la médula ósea, células precursoras de las plaquetas. En los pacientes, la función de la enzima UDP-Galactosa 4-Epimerasa es muy reducida, por lo que no se produce la glicosilación de proteínas esenciales para la formación de las plaquetas, expresándose en su forma inmadura y no funcional, que justifica su reducido recuento en sangre y el incremento del sangrado.
Estos hallazgos demuestran el papel esencial de GALE en la formación de proteínas maduras, y abre un nuevo horizonte para su estudio en otros órganos, tales como las neuronas, los miocitos o los hepatocitos, para comprender y expandir las alteraciones producidas por defectos de glicosilación a nivel sistémico.
Este estudio se incluye en la tesis doctoral internacional de Ana Marin Quílez, y han participado múltiples miembros del grupo Citogénetica del Hospital Universitario de Salamanca - IBSAL, dirigido por el Profesor Jesús María Hernández Rivas, e investigadores de la Universidad de Pavía (Italia), del Centro de Diagnóstico de Enfermedades Moleculares del CIBERER, del Hospital Clínico de Valladolid y del Hospital Virgen del Puerto de Plasencia. El estudio se enmarca en el proyecto multicéntrico nacional del Grupo Español de Alteraciones Plaquetarias Congénitas (GEAPC) de la Sociedad Española de Trombosis y Hemostasia (SETH).
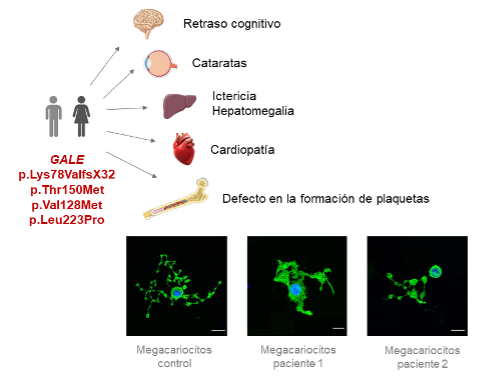
Explicación gráfica del estudio.
Artículo de referencia:
Marín-Quílez A; Di Buduo CA; Díaz-Ajenjo L; Abbonante V; Vuelta E; Soprano PM; Miguel-García C; Santos-Mínguez S; Serramito-Gómez I; Ruiz-Sala P; Peñarrubia MJ; Pardal E; Hernández-Rivas JM; González-Porras JR; García-Tuñón I; Benito R; Rivera J; Balduini A; Bastida JM. Novel variants in GALE cause syndromic macrothrombocytopenia by disrupting glycosylation and thrombopoiesis. Blood. 2022 Nov 17;blood. 2022016995. DOI: 10.1182/blood.2022016995




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




